22.2: Introduction to Energy Bands
- Page ID
- 46331
")
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)When two valence electron atomic orbitals in a simple molecule such as hydrogen combine to form a chemical bond, two possible molecular orbitals result. One molecular orbital is lowered in energy relative to the sum of the energies of the individual electron orbitals, and is referred to as the 'bonding' orbital. The other molecular orbital is raised in energy relative to the sum of the energies of the individual electron orbitals and is termed the 'anti-bonding' orbital.
In a solid, the same principles apply. If N valence electron atomic orbitals, all of the same energy, are taken and combined to form bonds, N possible energy levels will result. Of these, N/2 will be lowered in energy and N/2 will be raised in energy with respect to the sum of the energies of the N valence electron atomic orbitals.
However, instead of forming N/2 bonding levels all of the exact same energy, the allowed energy levels will be smeared out into energy bands. Within these energy bands local differences between energy levels are extremely small. The energy differences between the levels within the bands are much smaller than the difference between the energy of the highest bonding level and the energy of the lowest anti-bonding level. Like molecular orbitals, and also atomic orbitals, each energy level can contain at most two electrons of opposite spin.
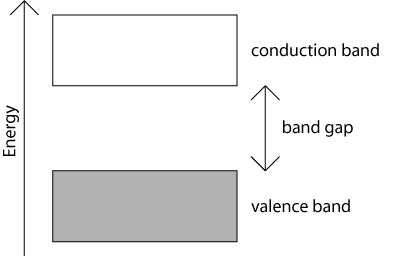
The allowed energy levels are so close together that they are sometimes considered as being continuous. It is very important to bear in mind that, while this is a useful and reasonable approximation in some calculations, the bands are actually composed of a finite number of very closely spaced electron energy levels.
If there is one electron from each atom associated with each of the N orbitals that are combined to form the bands, then because each resulting energy level can be doubly occupied, the 'bonding' band, or valence band will be completely filled and the 'anti-bonding' band, or conduction band will be empty. This is depicted schematically in the picture above by the grey shading of the valence band.
Electrons cannot have any values of energy that lie outside these bands. An electron can only move ('be promoted') from the valence band to the conduction band if it is given an energy at least as great as the band gap energy. This can happen if, for example, the electron were to absorb a photon of sufficiently high energy.
If, as in the above one-dimensional schematic, a band is completely filled with electrons, and the band immediately above it is empty, the material has an energy band gap. This band gap is the energy difference between the highest occupied state in the valence band and the lowest unoccupied state in the conduction band. The material is either a semiconductor if the band gap is relatively small, or an insulator if the band gap is relatively large.
Electrons in metals are also arranged in bands, but in a metal the electron distribution is different - electrons are not localised on individual atoms or individual bonds. In a simple metal with one valence electron per atom, such as sodium, the valence band is not full, and so the highest occupied electron states lie some distance from the top of the valence band. Such materials are good electrical conductors, because there are empty energy states available just above the highest occupied states, so that electrons can easily gain energy from an applied electric field and jump into these empty energy states.
The distinction between an insulator and a semiconductor is less precise. In general, a material with a band gap of less than about 3 eV is regarded as a semiconductor. A material with a band gap of greater than 3 eV will commonly be regarded as an insulator. A number of ceramics such as silicon carbide (SiC), titanium dioxide (TiO2), barium titanate (BaTiO3) and zinc oxide (ZnO) have band gaps around 3 eV and are regarded by ceramicists as semiconductors. Such ceramics are often referred to as wide-band-gap semicondutors.
The distinction between semiconductors and insulators arises because in small band gap materials at room temperature a small, but appreciable, number of electrons can be excited from the filled valence bands into the unfilled conduction bands simply by thermal vibration. This leads to semiconducting materials having electrical conductivities between those of metals and those of insulators.
The picture we have sketched here is only a very simple qualitative picture of the electronic structure of a semiconductor designed to capture essential aspects of the band structure in semiconductors relevant to this TLP. More precise and quantitative approaches exist - see Going Further. Such quantitative approaches are generally quite complex and require an understanding of quantum mechanics. Fortunately, the very simple qualitative picture described above for semiconductors is all that we need to be able to build upon and develop in this TLP.
An extension of the simple band energy diagram with only the vertical axis labelled as energy, with the horizontal axis unlabelled, is to plot the energy vertically against wave vector, k. From de Broglie's relationship p = k where p is momentum and is Planck's constant, h, divided by 2π. Such plots therefore relate energy to momentum. The reason why such plots are useful lies in the more quantitative methods referred to above, from which we shall simply quote useful results.
The energy of a classical, non-quantum, particle is proportional to the square of its momentum. This is also true for a free electron, as in the most simple picture possible of valence electrons in metals where the electrostatic potential from the nuclei is ignored. However, in a real crystalline solid the periodicity of the lattice and the electrostatic potential from the nuclei together mean that in the quantum world in a crystalline material the electron energy, E, is not simply proportional to the square of the momentum, and so is not proprtional to the square of the wave vector, k.
In these E-k diagrams, often called band diagrams, plotted in what is referred to as a reduced zone scheme, the momentum that is plotted is actually a quantity called crystal momentum. The distinction between momentum and crystal momentum arises from the periodicity of the solid. Fortunately, this distinction is not important for understanding this TLP on semiconductors.
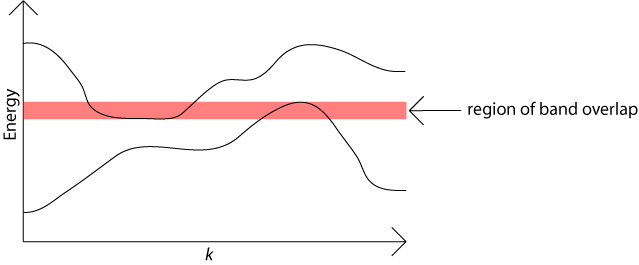
There are usually many different values of electron energy possible for any given value of the electron momentum. Each possible energy value lies in one of the energy bands.
When plotted against the wave vector, k, the bands of allowed energy are not really flat. This means that bands can overlap in energy, as the maximum value in one band may be higher then the minimum value in another band. In this case the relevant maximum and minimum will occur for different values of k because energy bands never cross over each other. This is one way in which metals can have partially filled energy bands. The available energy states are filled with electrons starting with those lowest in energy. Such overlapping of bands as a function of k does not occur in semiconductors.